Rod dystrophy secondary to PRPF31 mutation
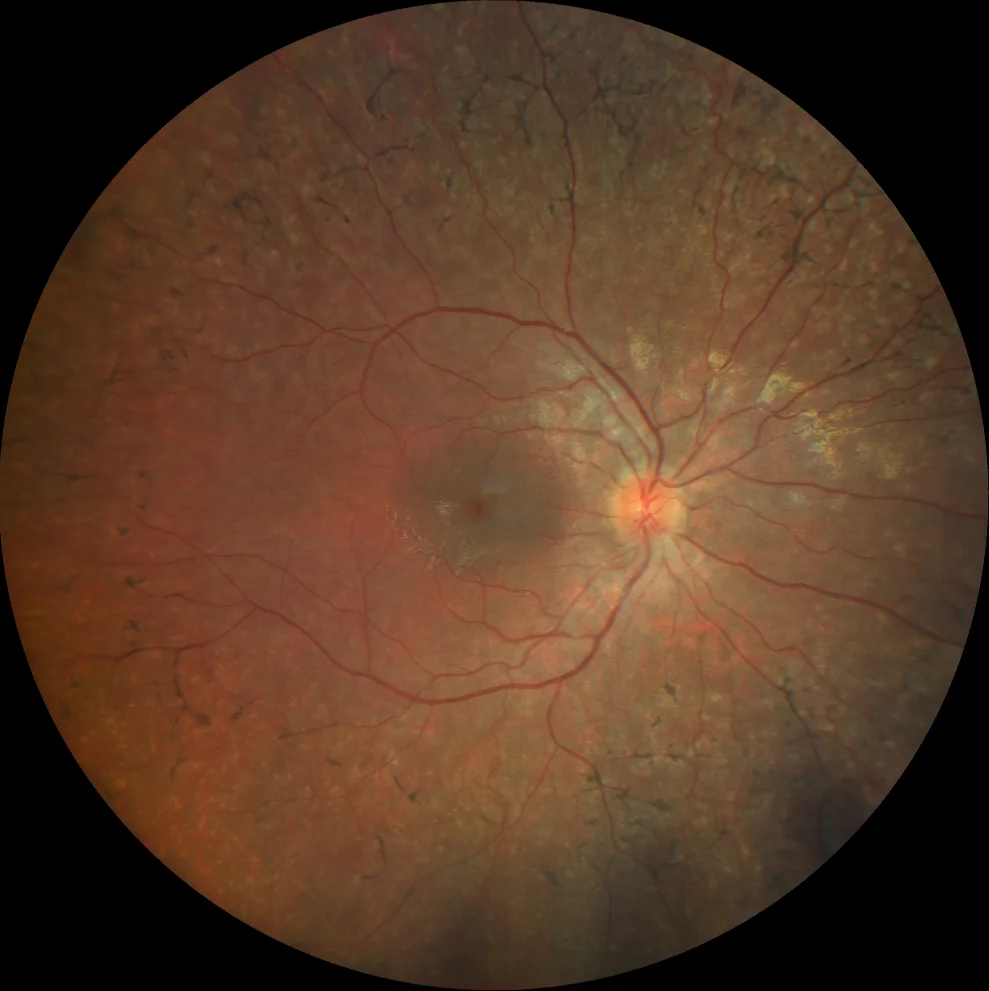
A and B. Color fundusography (Clarus 500, Carl Zeiss Meditec ASG, Jena, Germany) of the older sister's right and left eyes, showing midperipheral bone spicules and vascular attenuation.
A and B. Color fundusography (Clarus 500, Carl Zeiss Meditec ASG, Jena, Germany) of the older sister's right and left eyes, showing midperipheral bone spicules and vascular attenuation.
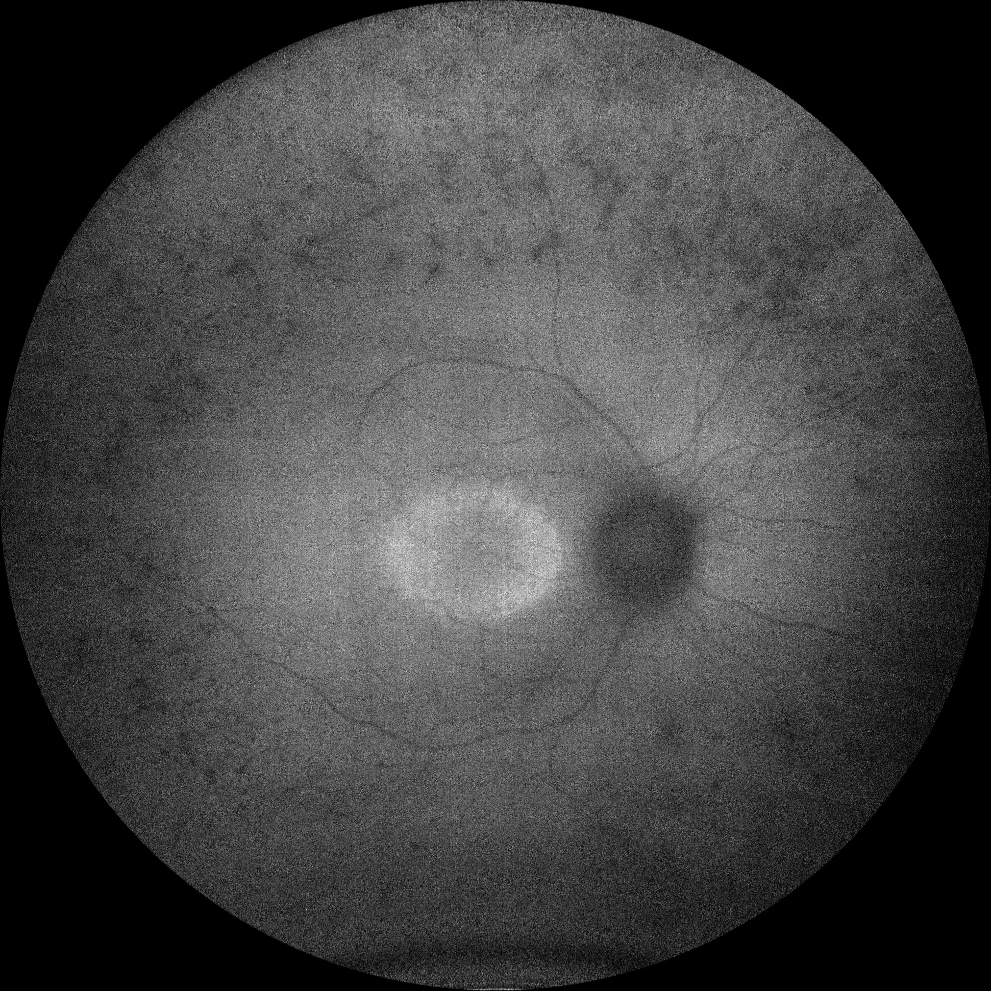
C and D. Autofluorescence (Clarus 500, Carl Zeiss Meditec ASG, Jena, Germany) of the right and left eyes of the older sister, showing hypoautofluorescence in the bone spicules in the periphery, as well as a hyperautofluorescent ring in the macula in both eyes.
C and D. Autofluorescence (Clarus 500, Carl Zeiss Meditec ASG, Jena, Germany) of the right and left eyes of the older sister, showing hypoautofluorescence in the bone spicules in the periphery, as well as a hyperautofluorescent ring in the macula in both eyes.
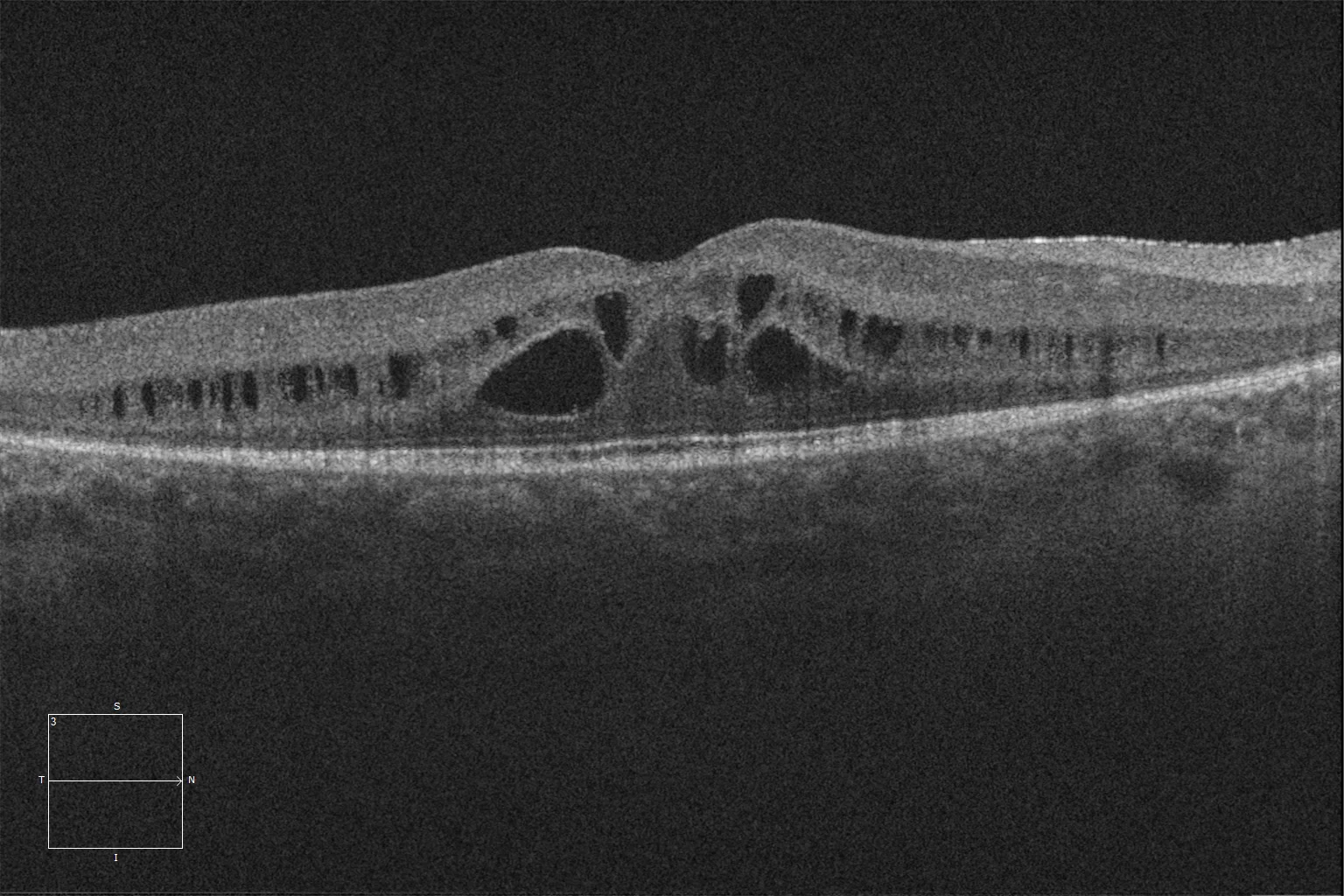
E and F. Optical coherence tomography (Cirrus 5000, Carl Zeiss Meditec ASG, Jena, Germany) of the macular degeneration of the right and left eyes of the older sister, showing retinal schisis, preservation of the ellipsoid and ELM layers in the central retina and their loss as we go beyond the area corresponding to the hyperautofluorescent ring.
E and F. Optical coherence tomography (Cirrus 5000, Carl Zeiss Meditec ASG, Jena, Germany) of the macular degeneration of the right and left eyes of the older sister, showing retinal schisis, preservation of the ellipsoid and ELM layers in the central retina and their loss as we go beyond the area corresponding to the hyperautofluorescent ring.
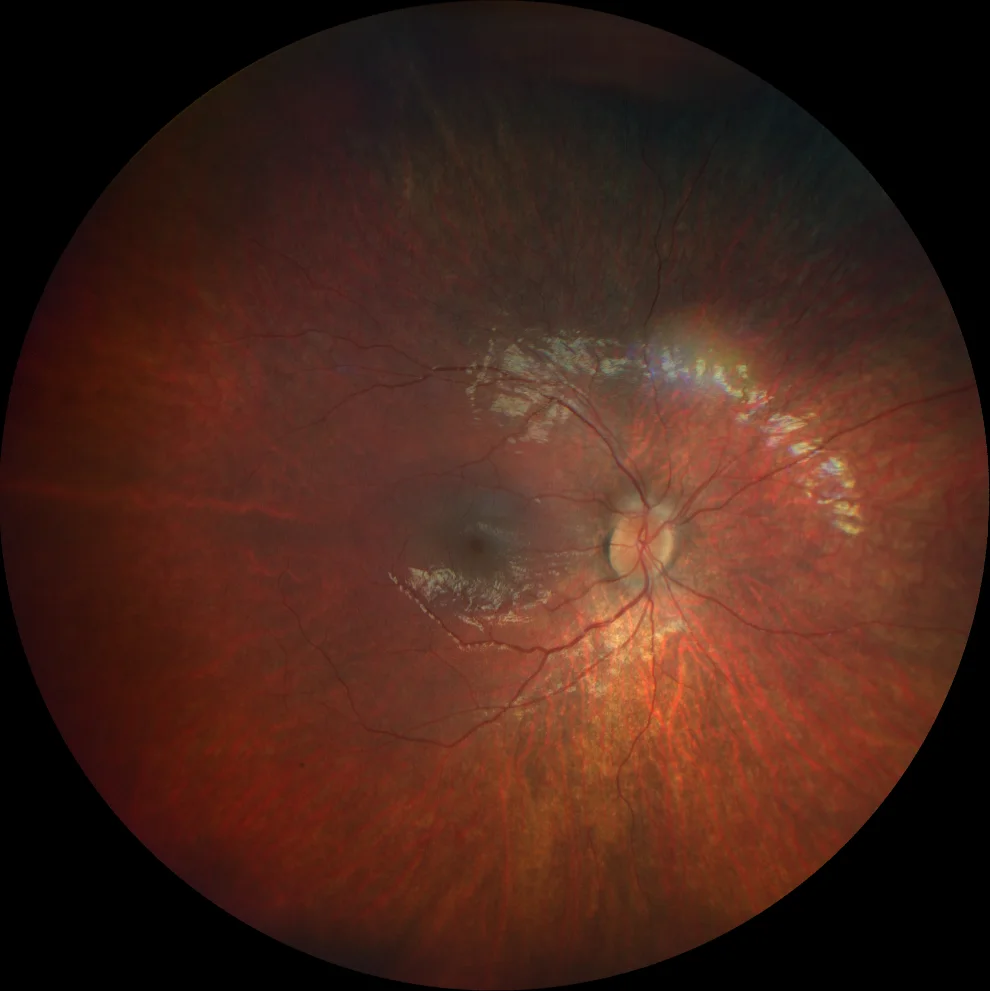
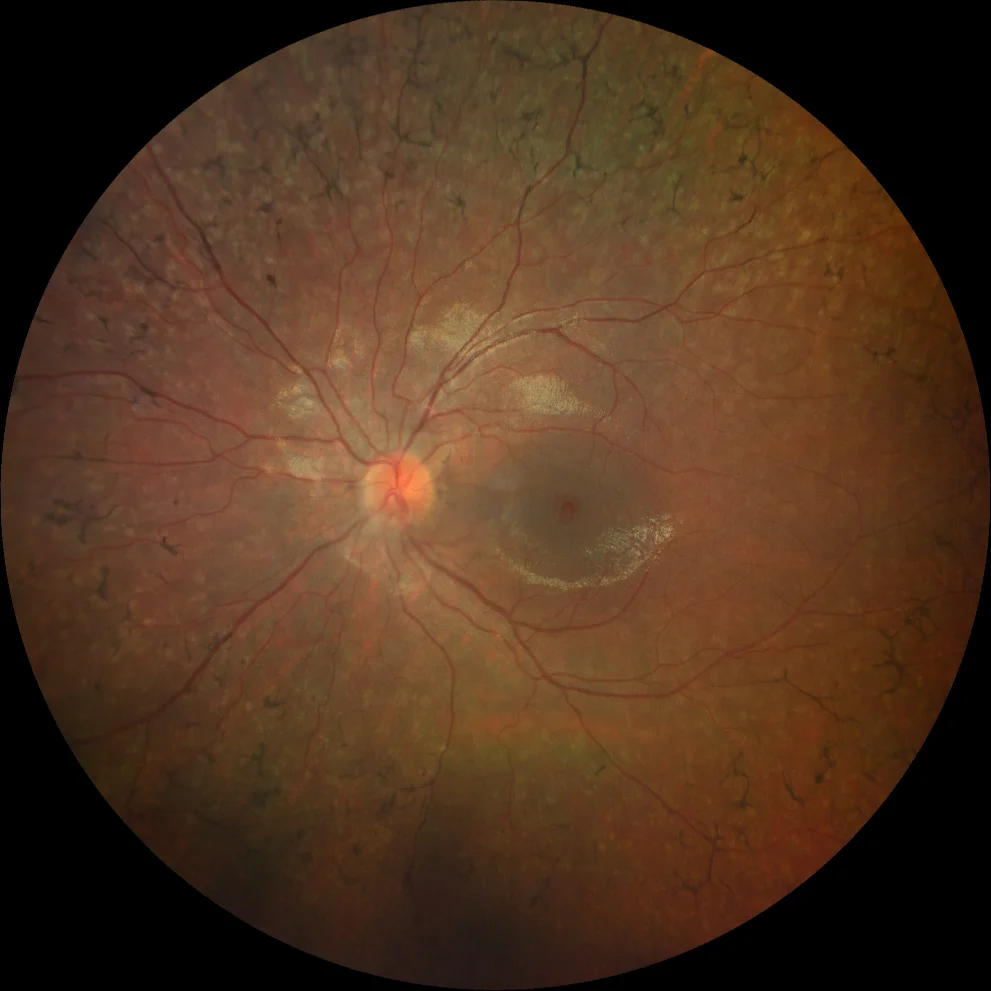
G and H. Color fundusography (Clarus 500, Carl Zeiss Meditec ASG, Jena, Germany) of the right and left eyes of the younger sister, showing mild vascular attenuation and absence of bone spicules.
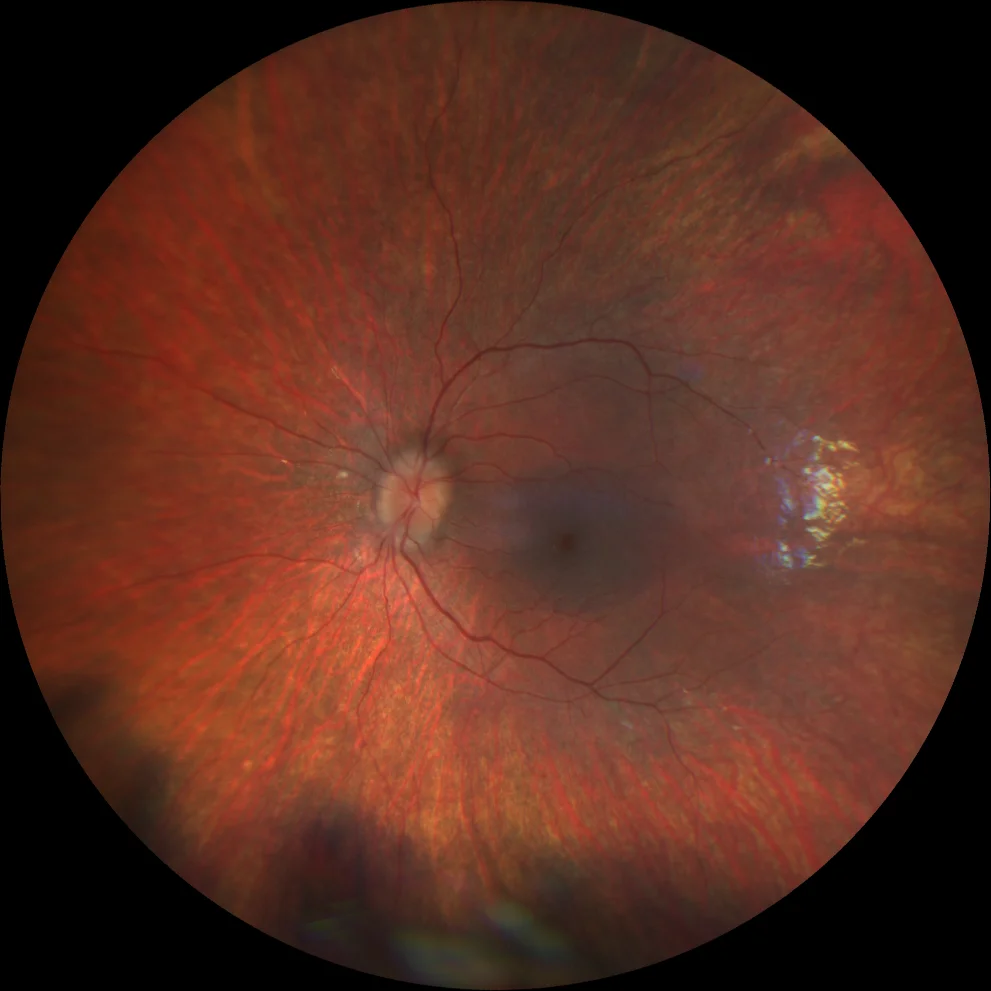
G and H. Color fundusography (Clarus 500, Carl Zeiss Meditec ASG, Jena, Germany) of the right and left eyes of the younger sister, showing mild vascular attenuation and absence of bone spicules.
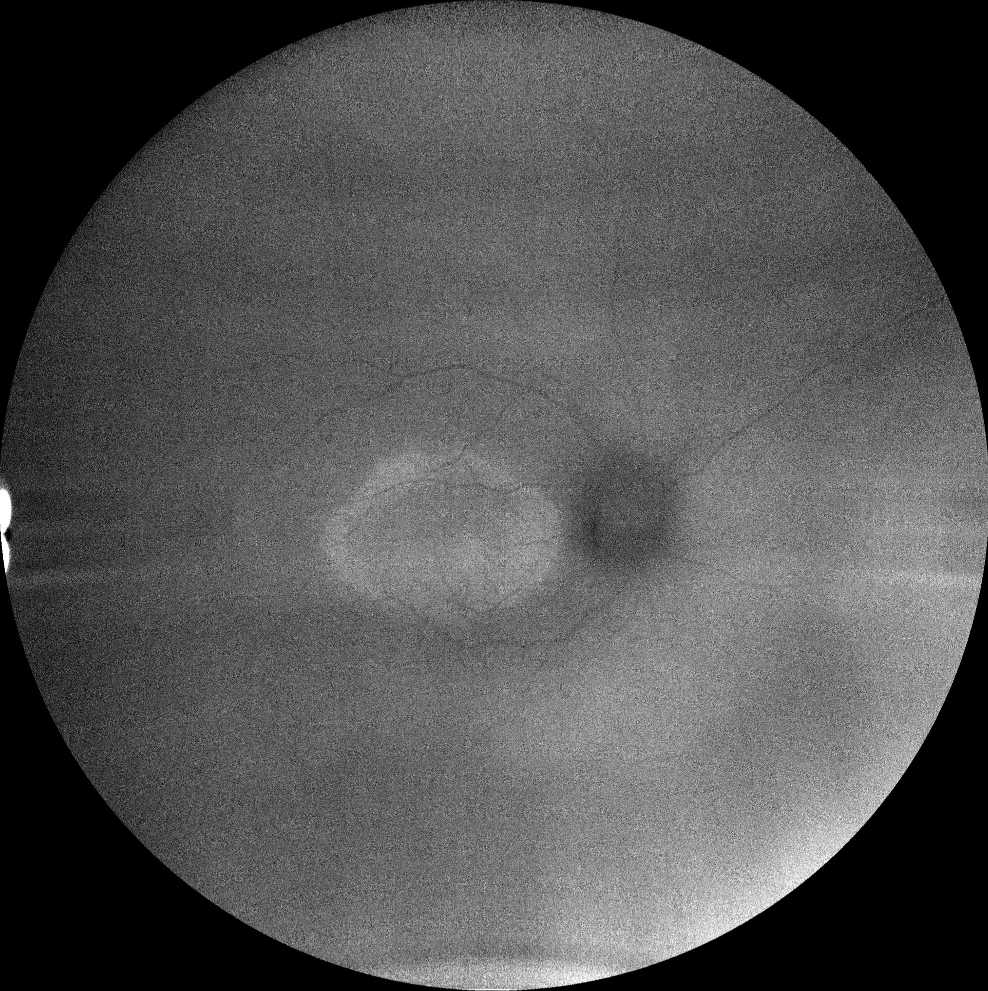
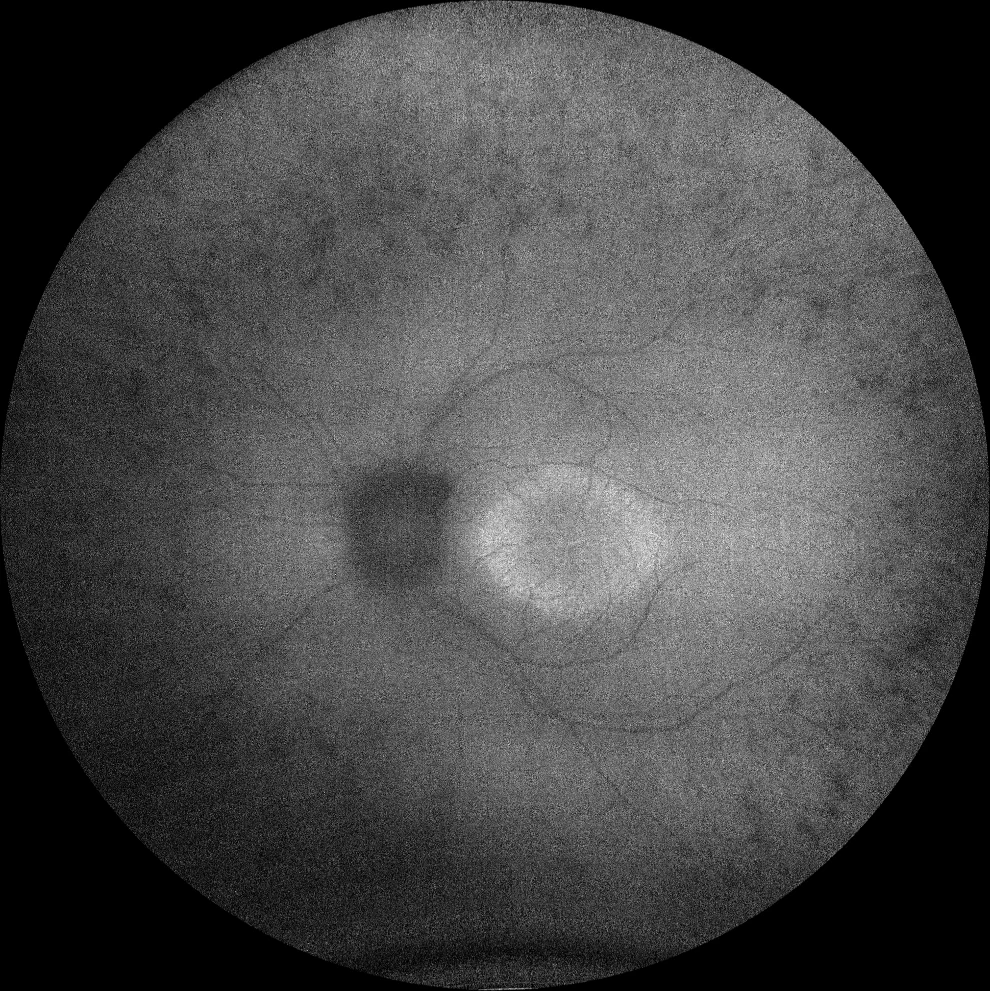
I and J. Autofluorescence (Clarus 500, Carl Zeiss Meditec ASG, Jena, Germany) of the right and left eyes of the little sister, showing the hyperautofluorescent ring in the macula in both eyes.
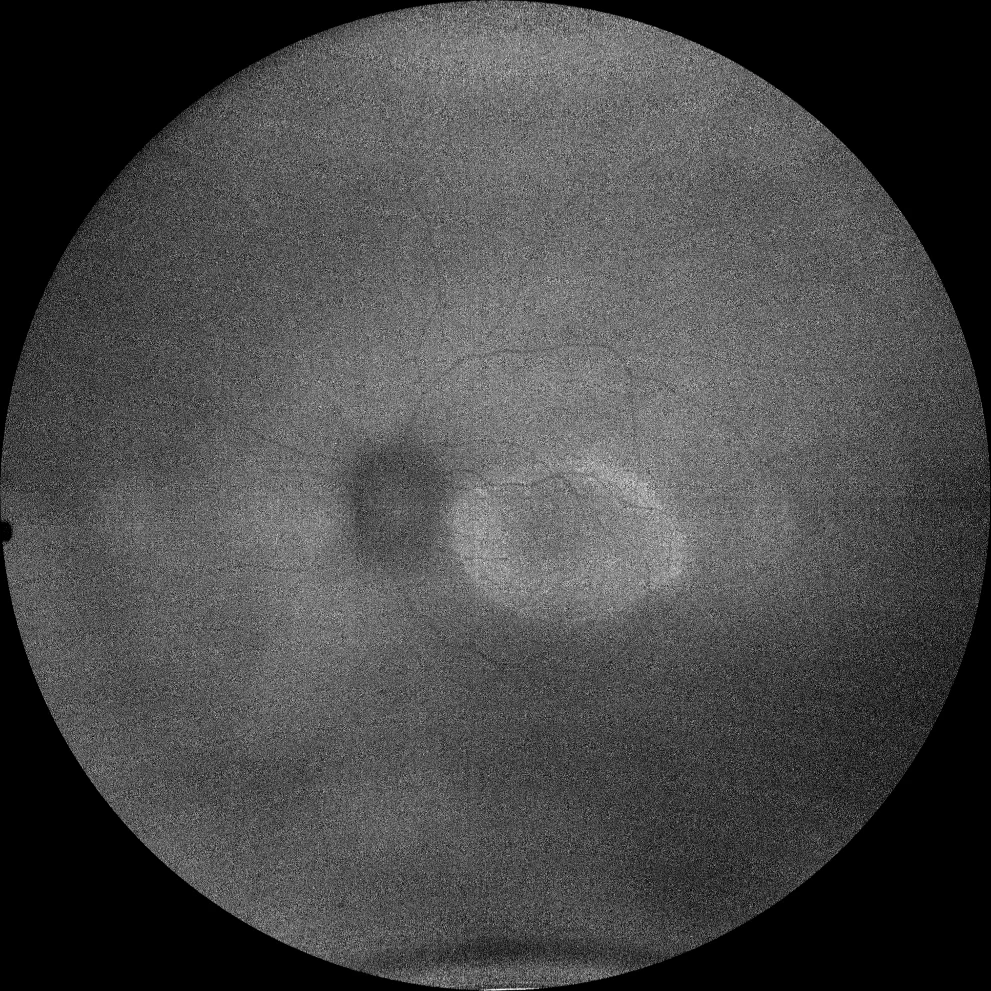
I and J. Autofluorescence (Clarus 500, Carl Zeiss Meditec ASG, Jena, Germany) of the right and left eyes of the little sister, showing the hyperautofluorescent ring in the macula in both eyes.
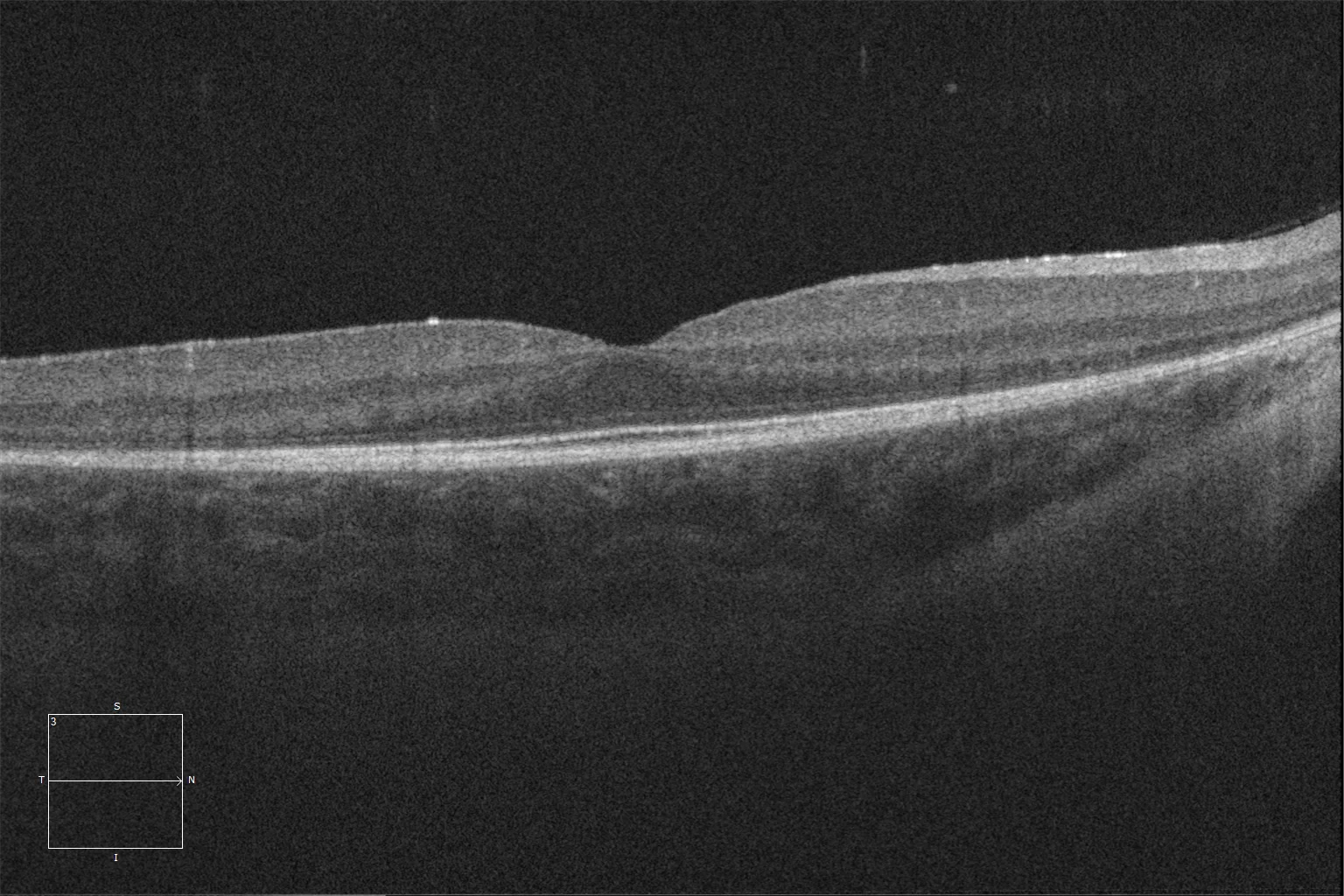
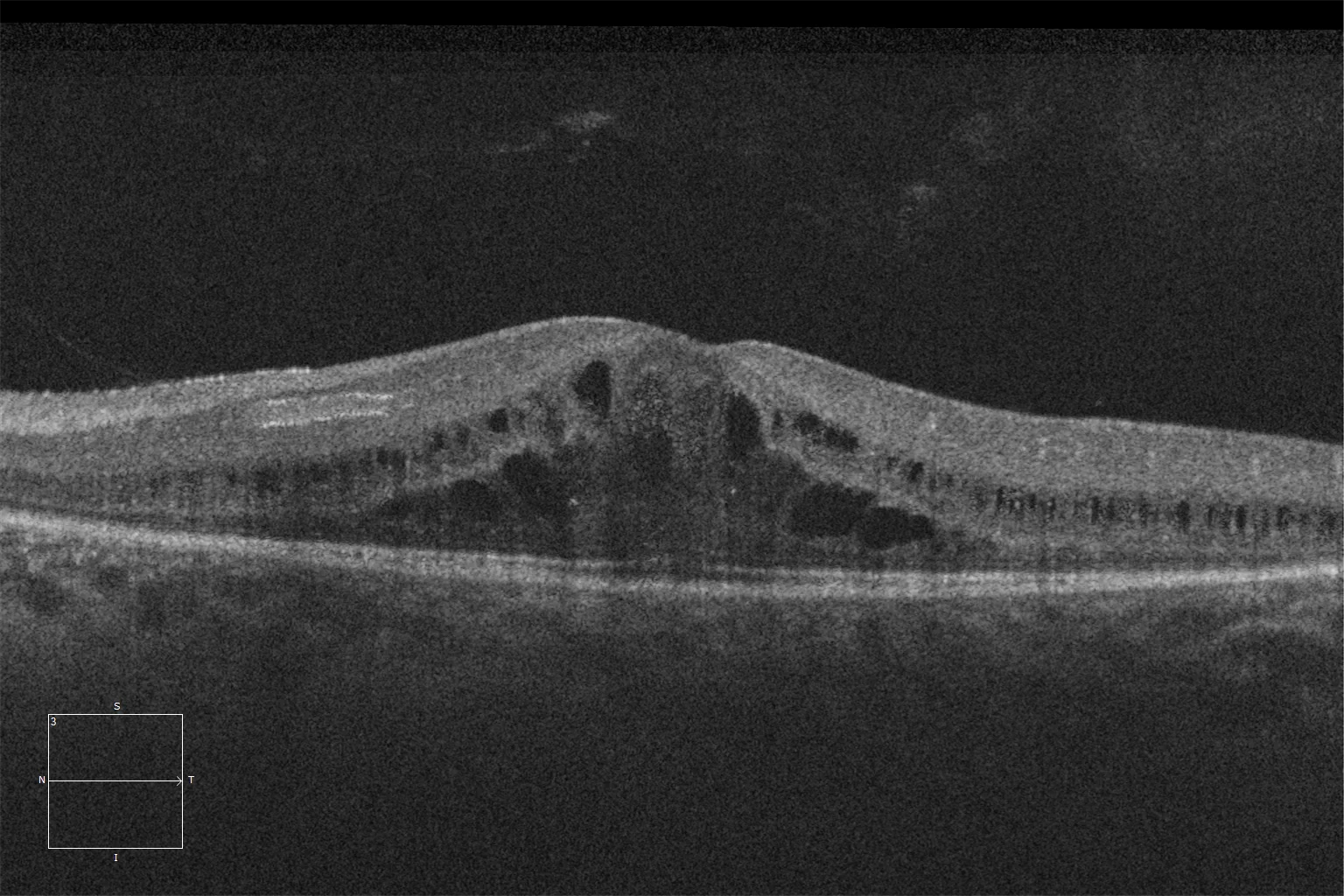
K and L. Optical coherence tomography (Cirrus 5000, Carl Zeiss Meditec ASG, Jena, Germany) of the macular region of the right and left eyes of the younger sister, showing preservation of the ellipsoid and ELM layers in the central retina and their loss as we go beyond the area corresponding to the hyperautofluorescent ring.
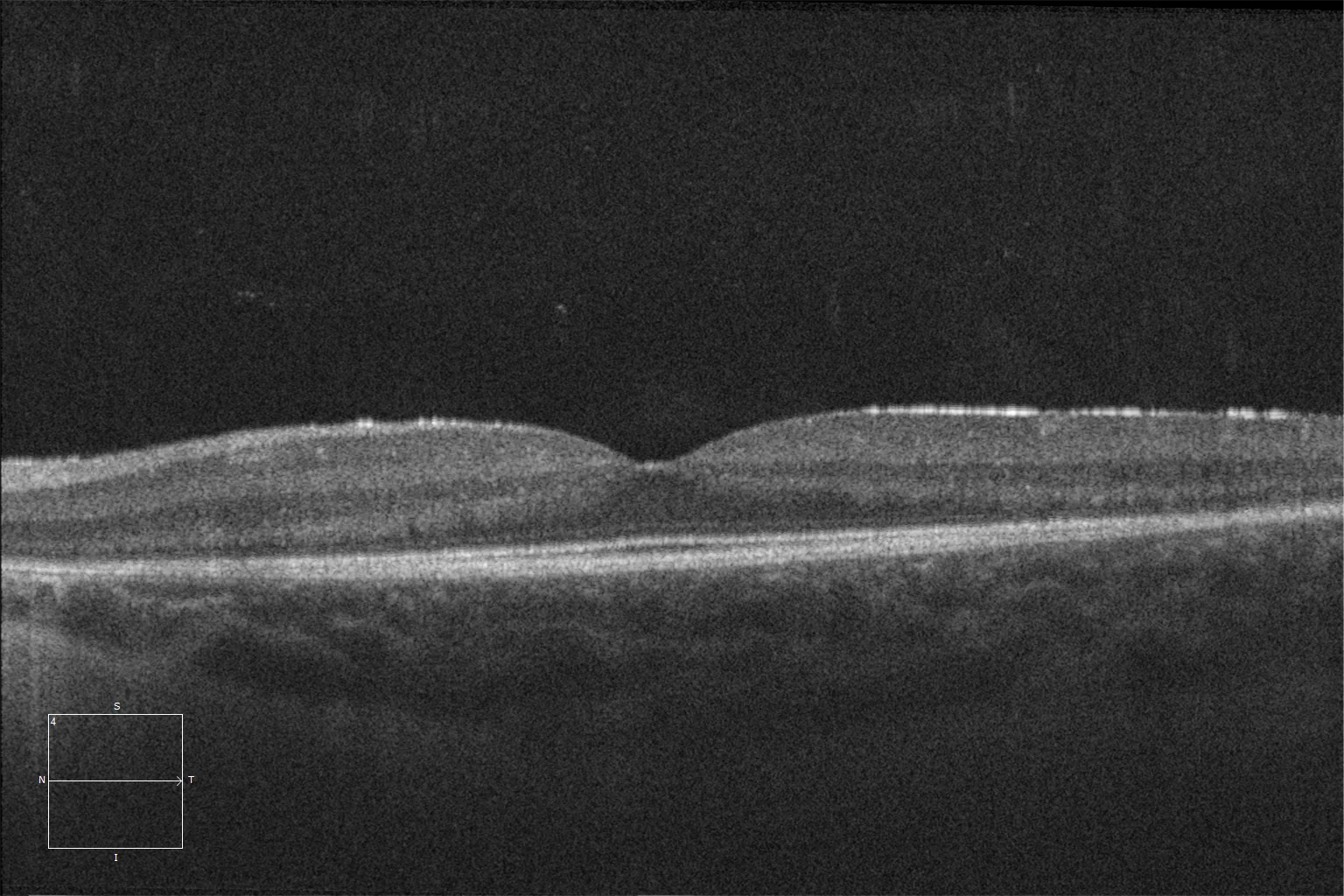
K and L. Optical coherence tomography (Cirrus 5000, Carl Zeiss Meditec ASG, Jena, Germany) of the macular region of the right and left eyes of the younger sister, showing preservation of the ellipsoid and ELM layers in the central retina and their loss as we go beyond the area corresponding to the hyperautofluorescent ring.
Description
Retinitis pigmentosa (also known as rod dystrophy) encompasses a group of clinically and genetically heterogeneous hereditary retinal disorders. It is characterized by progressive dysfunction of the retinal pigment epithelium and photoreceptors, affecting rods first and cones later. It usually manifests as difficulty with night vision and reduction of peripheral visual field, starting in adolescence. The classic clinical triad consists of arteriolar attenuation, retinal pigmentary changes in the mid-periphery (generally hyperpigmentation in the form of bone spicules), and waxy pallor of the optic disc. Classically, the electrophysiological response in the full-field electroretinogram includes a profound decrease or abolition of scotopic responses, along with a decrease in photopic response (which in advanced stages may also be abolished). Its prevalence is 1:3000 to 1:5000, and it can present in isolation – more frequently – or in association with other syndromic features. Given the high genotypic variability, autosomal dominant, autosomal recessive, and X-linked recessive inheritance patterns are possible. Even some mitochondrial inherited diseases can produce a retinitis pigmentosa phenotype, such as Kearns-Sayre syndrome. Isolated cases may also occur, sometimes due to de novo mutations.