Methylmalonic Acidemia with Homocystinuria Type CbLC
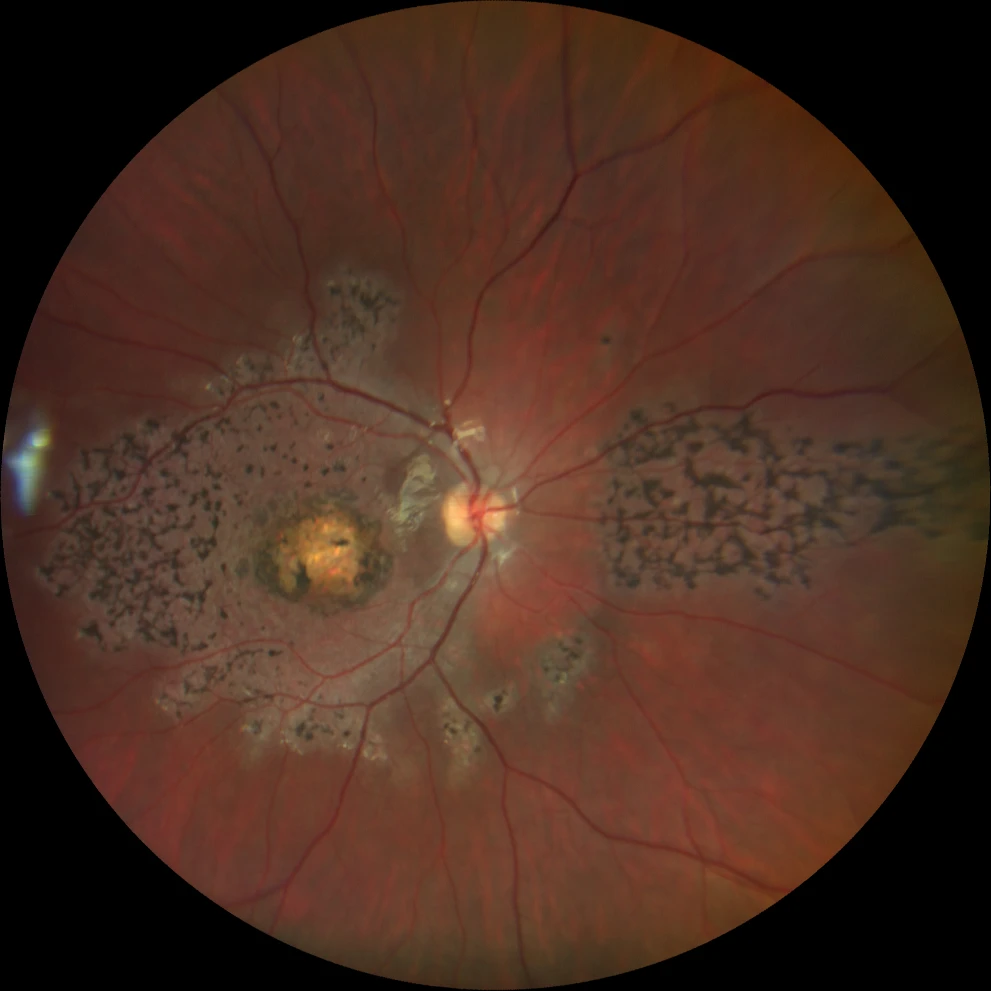
A and B. Color fundus photographs (Clarus 500, Carl Zeiss Meditec ASG, Jena, Germany) of the right and left eyes, showing bilateral bull's-eye macular atrophy, together with pigment in symmetrically distributed bone spicules in the macula and in the nasal retina.
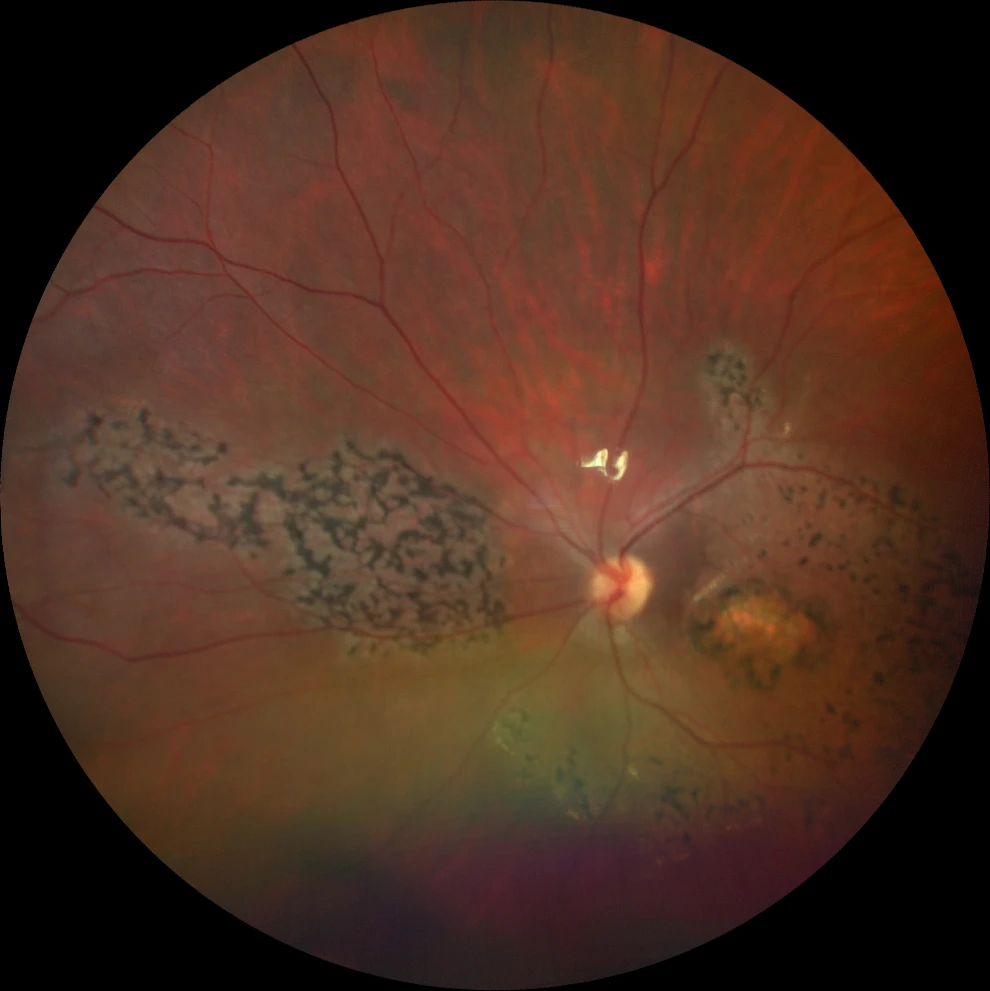
A and B. Color fundus photographs (Clarus 500, Carl Zeiss Meditec ASG, Jena, Germany) of the right and left eyes, showing bilateral bull's-eye macular atrophy, together with pigment in symmetrically distributed bone spicules in the macula and in the nasal retina.
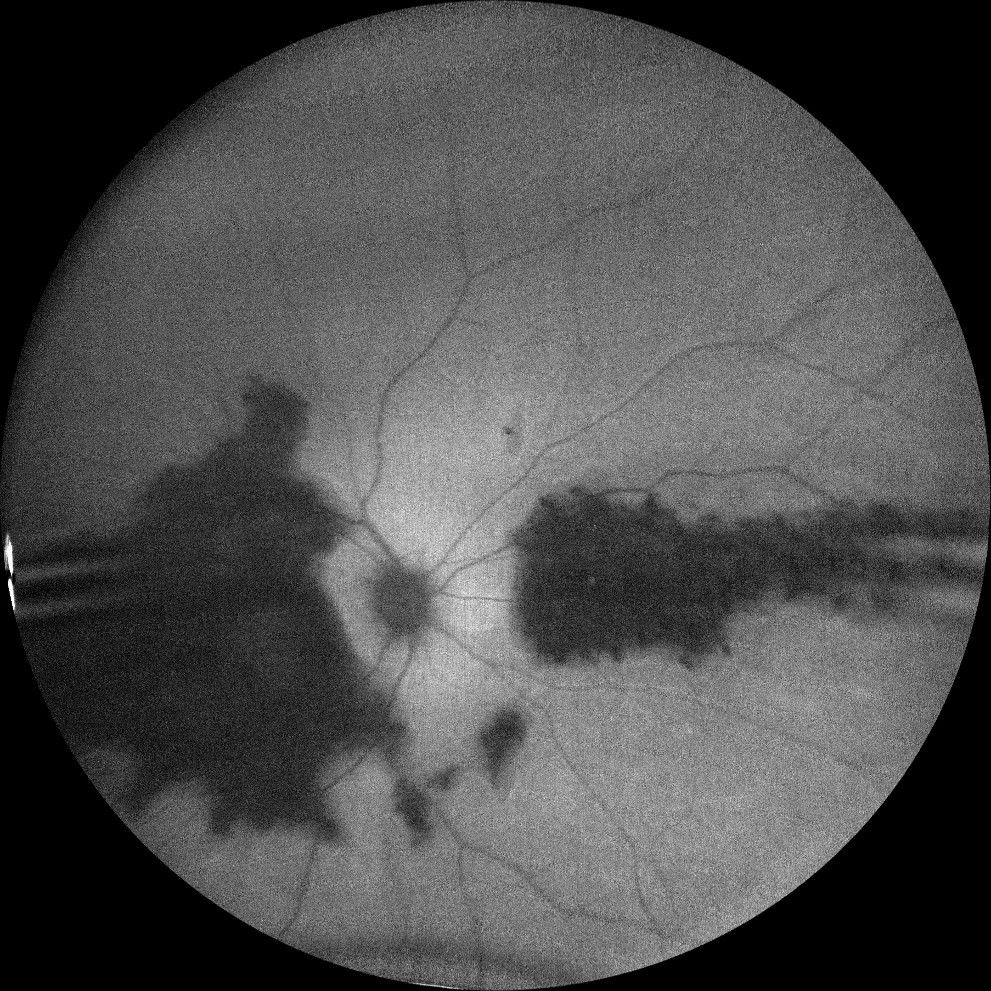
C and D. Autofluorescence images (Clarus 500, Carl Zeiss Meditec ASG, Jena, Germany) of the right and left eyes, showing macular hypoautofluorescence and the affected nasal retinal patch.
C and D. Autofluorescence images (Clarus 500, Carl Zeiss Meditec ASG, Jena, Germany) of the right and left eyes, showing macular hypoautofluorescence and the affected nasal retinal patch.
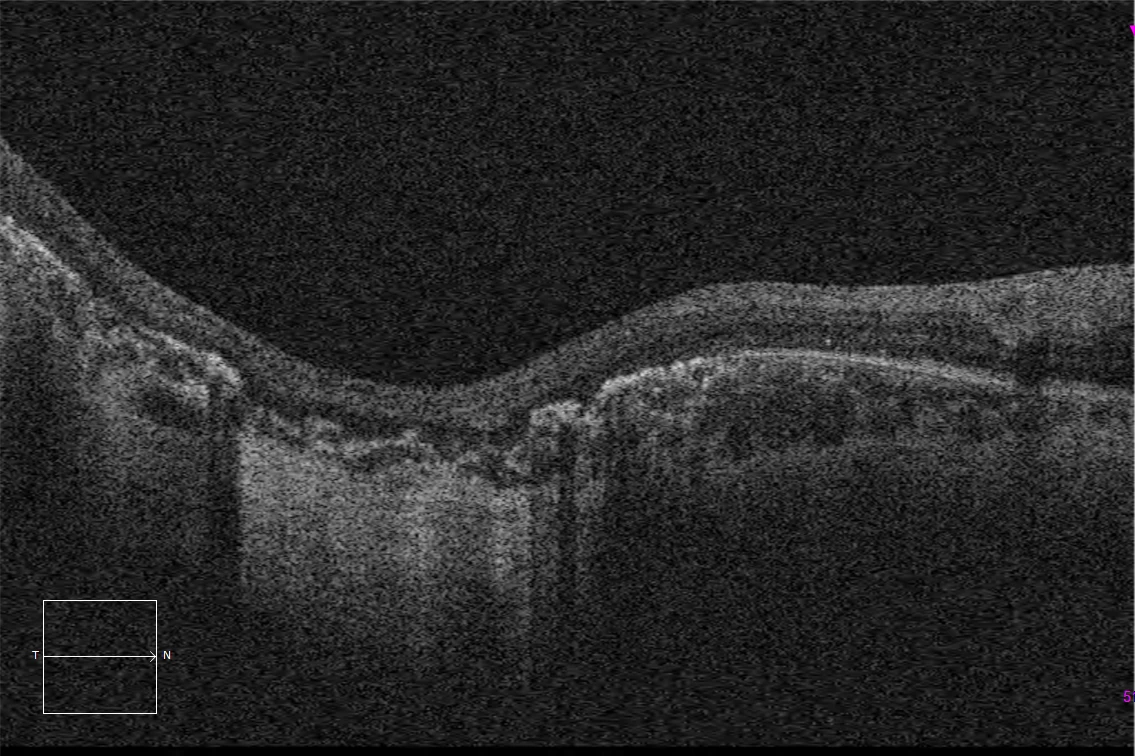
E and F. Macular HD optical coherence tomography (Cirrus 5000, Carl Zeiss Meditec ASG, Jena, Germany) of the right and left eyes, showing complete atrophy of the outer retina and subfoveal pigment epithelium, with preservation of these layers in the peripapillary retina.
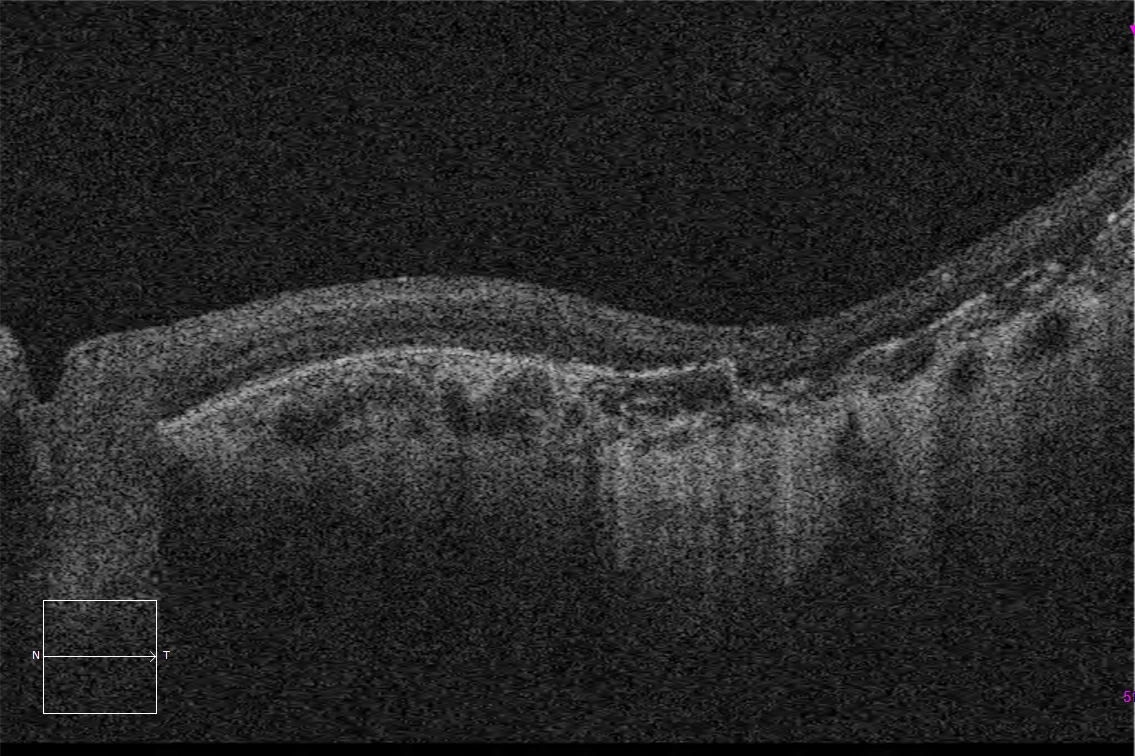
E and F. Macular HD optical coherence tomography (Cirrus 5000, Carl Zeiss Meditec ASG, Jena, Germany) of the right and left eyes, showing complete atrophy of the outer retina and subfoveal pigment epithelium, with preservation of these layers in the peripapillary retina.
Description
Methylmalonic acidemia with homocystinuria is a congenital error in metabolism, consisting of an inability to convert vitamin B12 (cobalamin) into its active forms, which are necessary for the conversion of homocysteine to methionine, as well as for proper mitochondrial activity. The absence of these processes will lead to an accumulation of homocysteine and its consequent urinary excretion (homocystinuria) as well as acidemia resulting from mitochondrial inactivity (methylmalonic acidemia). The most commonly affected gene in this disease is MMACHC, causing methylmalonic acidemia with homocystinuria type CbLC.
The fundus examination revealed bilateral bull’s eye maculopathy as well as the appearance of bone spicules perimacular and nasal to the optic nerve, which were characteristically hypoautofluorescent in autofluorescence imaging. The electroretinogram showed a decrease in photopic response and an abolition of scotopic response.