Dystrophie des bâtonnets secondaire à une mutation du gène PRPF31
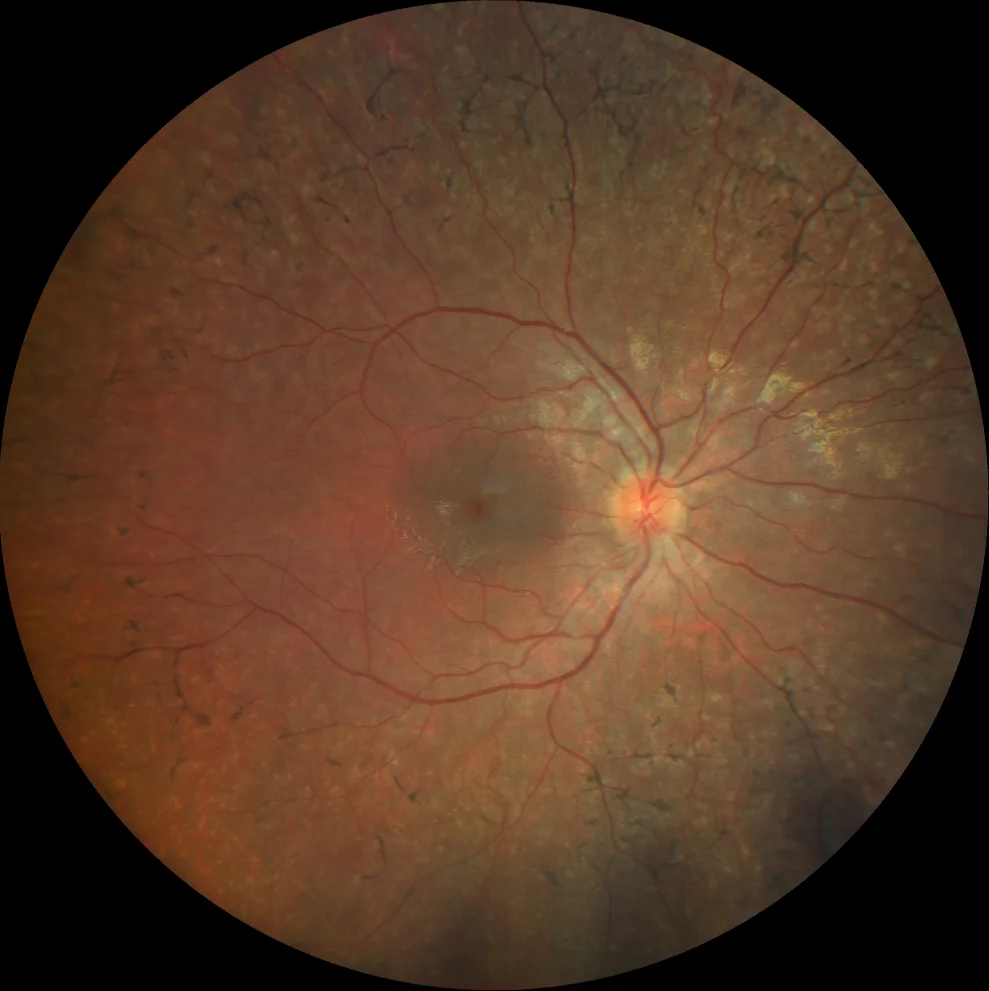
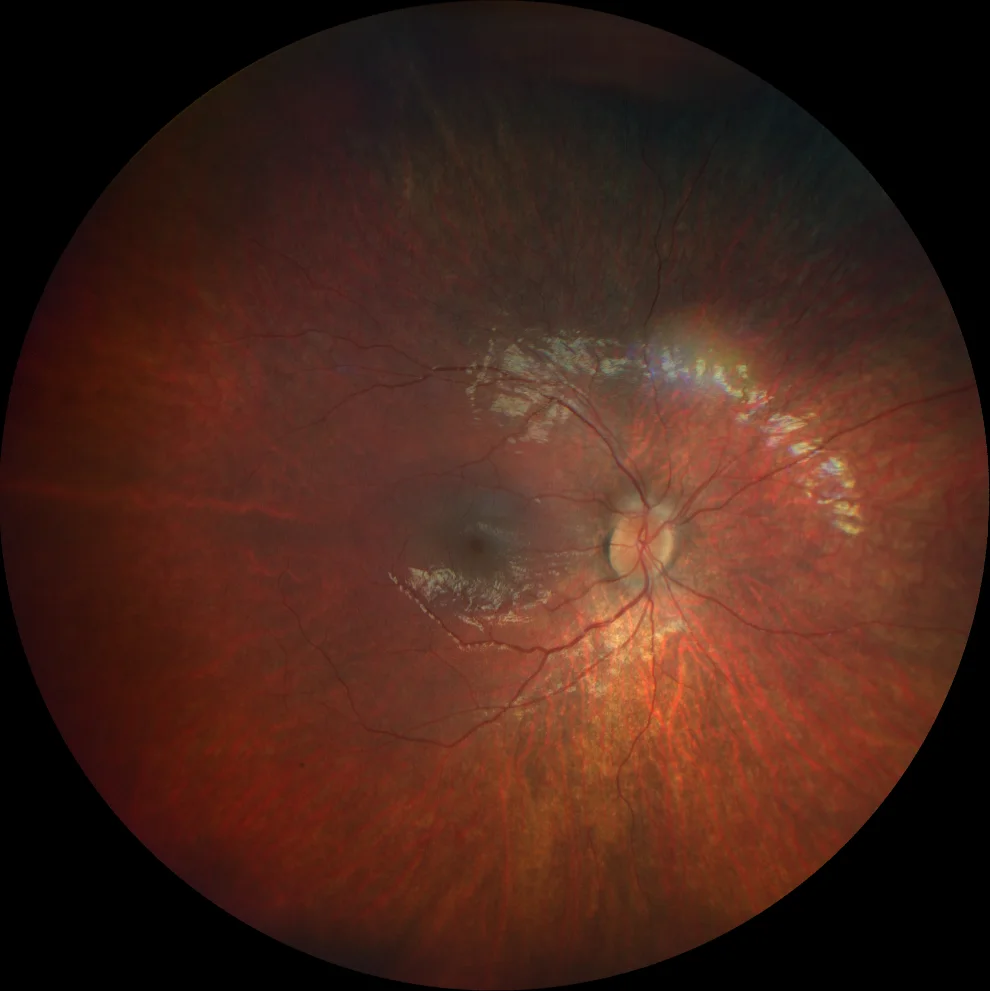
A et B. Rétinographie couleur (Clarus 500, Carl Zeiss Meditec ASG, Jena, Allemagne) des yeux droit et gauche de la sœur aînée, montrant des spicules osseux dans la périphérie moyenne et une atténuation vasculaire.
A et B. Rétinographie couleur (Clarus 500, Carl Zeiss Meditec ASG, Jena, Allemagne) des yeux droit et gauche de la sœur aînée, montrant des spicules osseux dans la périphérie moyenne et une atténuation vasculaire.
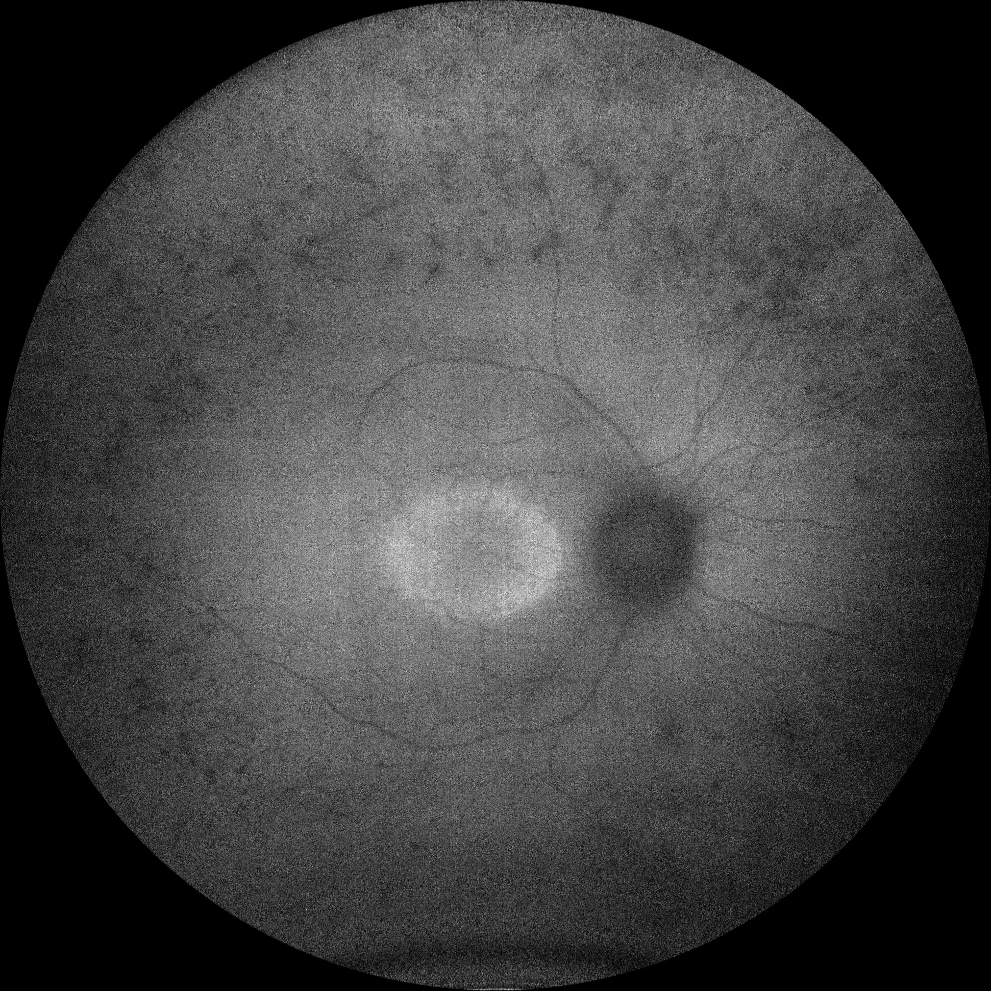
C et D. Autofluorescence (Clarus 500, Carl Zeiss Meditec ASG, Iéna, Allemagne) des yeux droit et gauche de la sœur aînée, montrant une hypoautofluorescence des spicules osseux en périphérie, ainsi qu'un anneau hyperautofluorescent dans la macula des deux yeux.
C et D. Autofluorescence (Clarus 500, Carl Zeiss Meditec ASG, Iéna, Allemagne) des yeux droit et gauche de la sœur aînée, montrant une hypoautofluorescence des spicules osseux en périphérie, ainsi qu'un anneau hyperautofluorescent dans la macula des deux yeux.
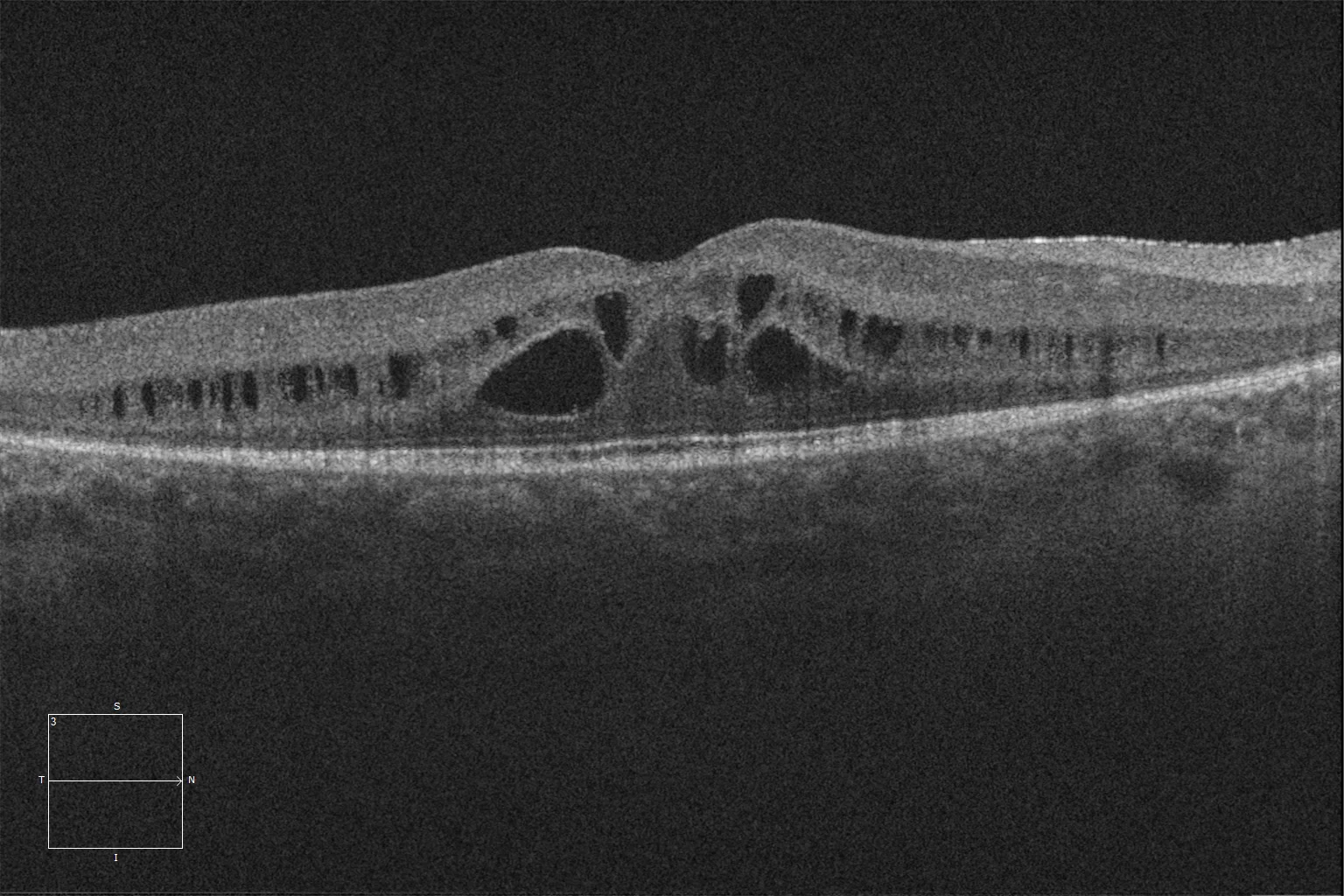
E et F. Tomographie par cohérence optique (Cirrus 5000, Carl Zeiss Meditec ASG, Iéna, Allemagne) : examen maculaire des yeux droit et gauche de la sœur aînée, montrant une schisis rétinienne, la préservation des couches ellipsoïde et ELM dans la rétine centrale et leur disparition au-delà de la zone correspondant à l’anneau hyperautofluorescent.
E et F. Tomographie par cohérence optique (Cirrus 5000, Carl Zeiss Meditec ASG, Iéna, Allemagne) : examen maculaire des yeux droit et gauche de la sœur aînée, montrant une schisis rétinienne, la préservation des couches ellipsoïde et ELM dans la rétine centrale et leur disparition au-delà de la zone correspondant à l’anneau hyperautofluorescent.
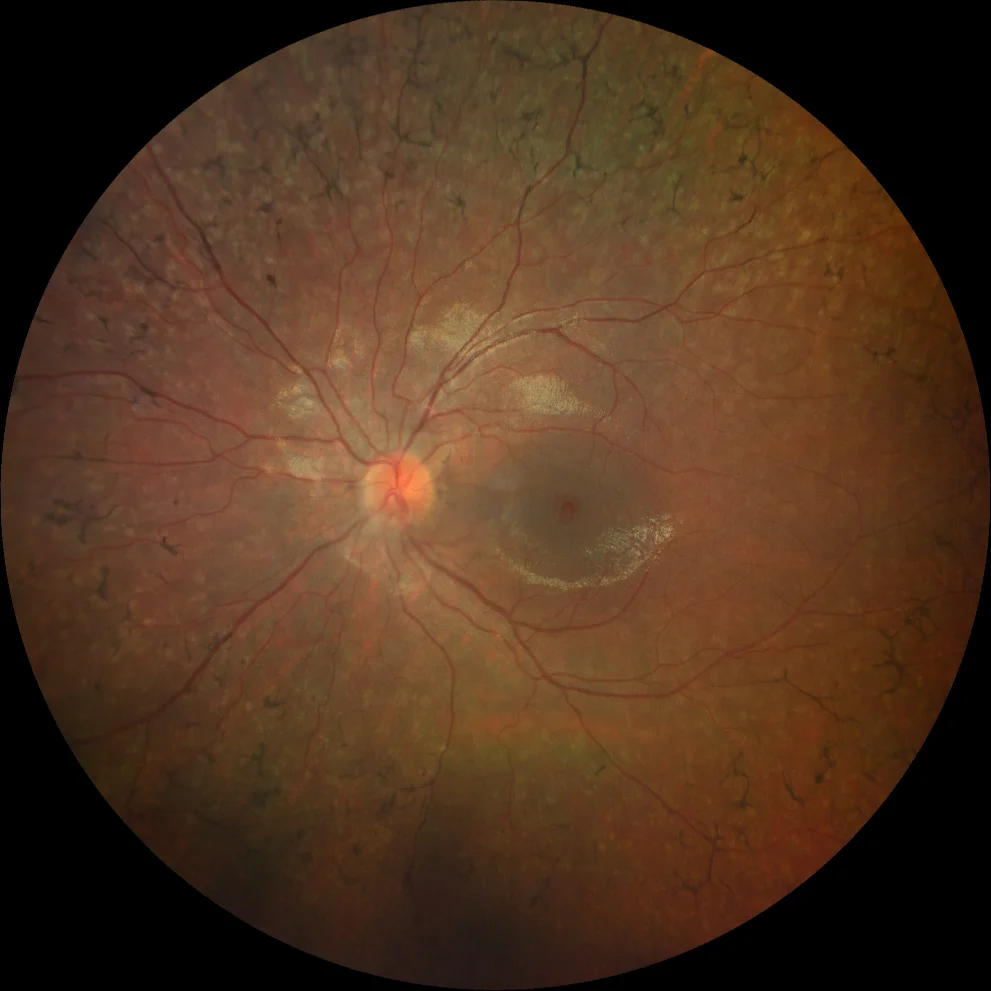
G et H. Rétinographie couleur (Clarus 500, Carl Zeiss Meditec ASG, Iéna, Allemagne) des yeux droit et gauche de la sœur cadette, montrant une légère atténuation vasculaire et l'absence de spicules osseux.
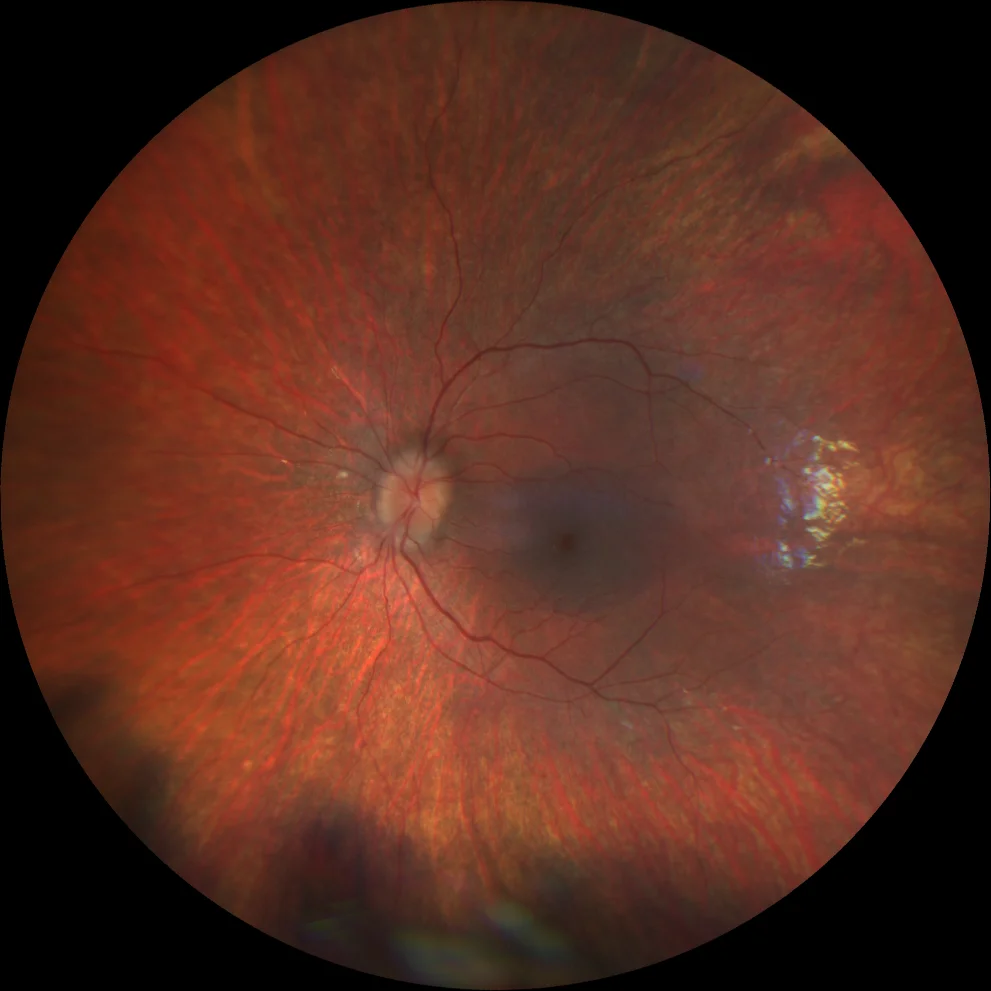
G et H. Rétinographie couleur (Clarus 500, Carl Zeiss Meditec ASG, Iéna, Allemagne) des yeux droit et gauche de la sœur cadette, montrant une légère atténuation vasculaire et l'absence de spicules osseux.
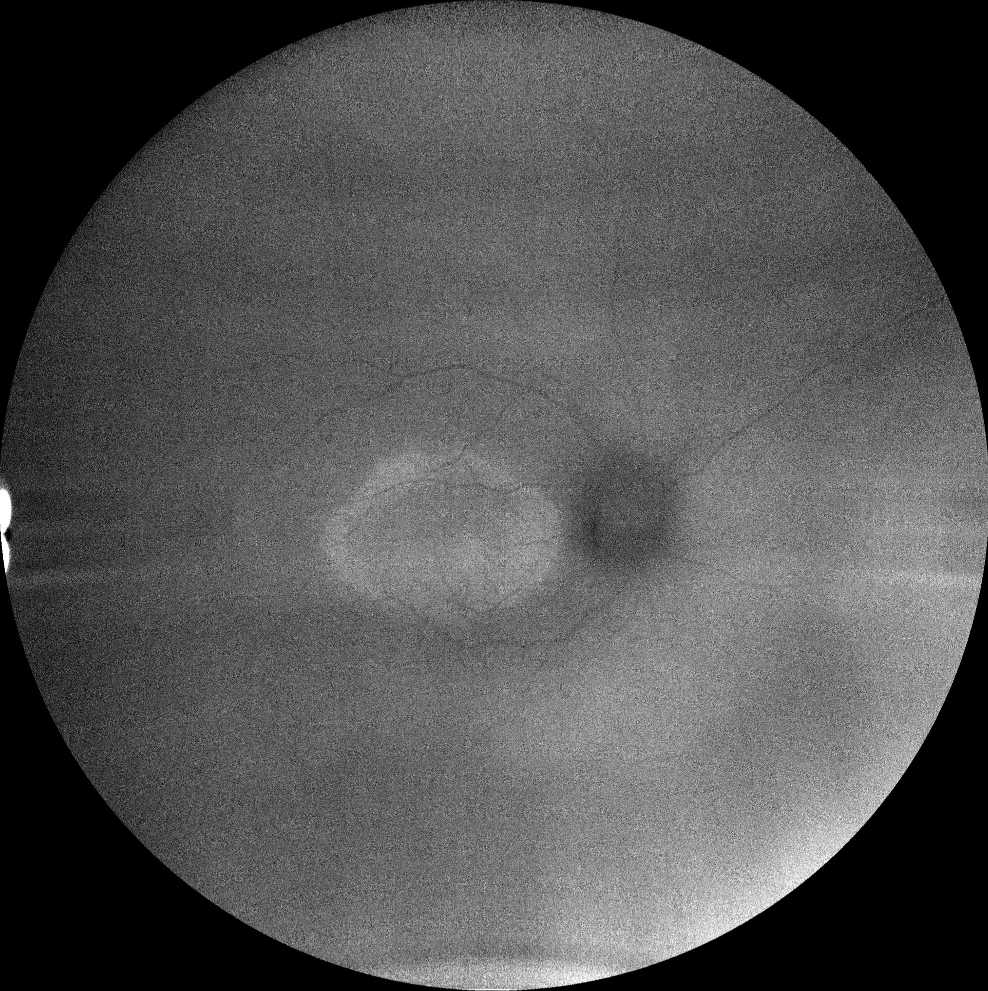
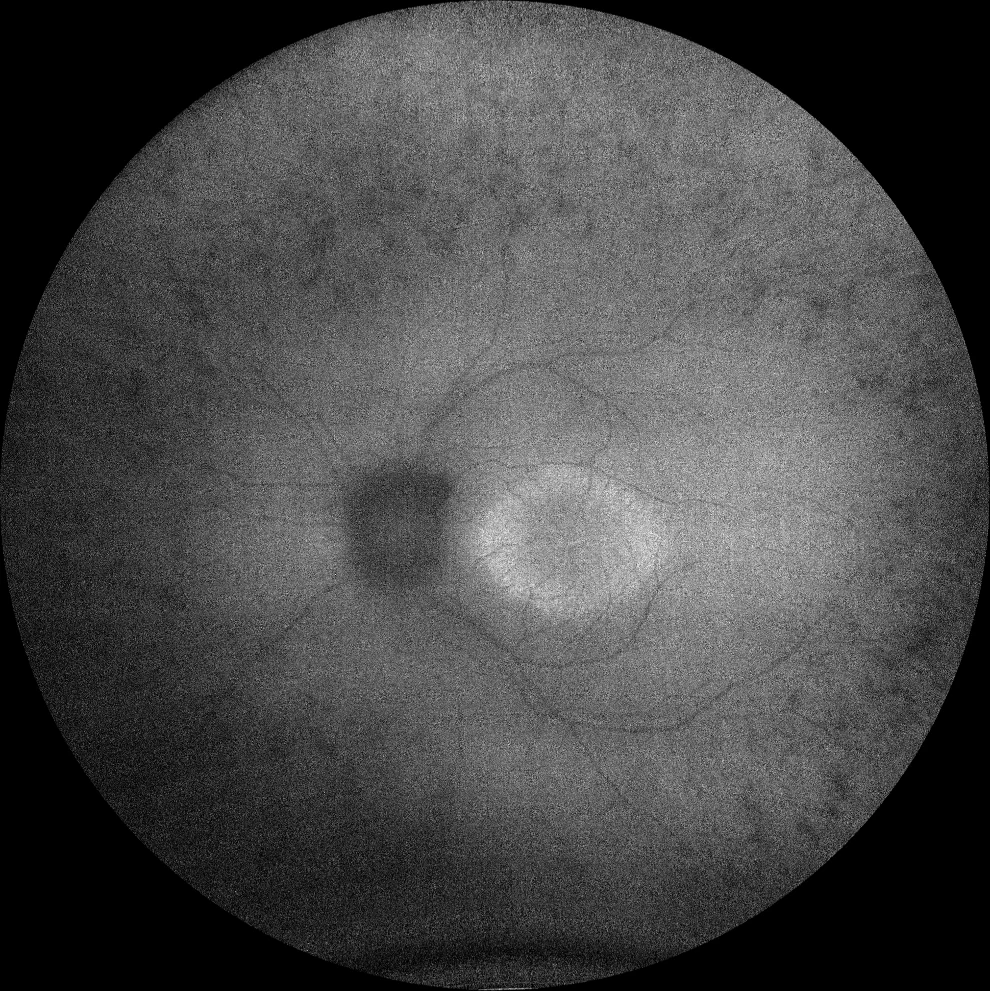
I et J. Autofluorescence (Clarus 500, Carl Zeiss Meditec ASG, Iéna, Allemagne) des yeux droit et gauche de la petite sœur, montrant l'anneau hyperautofluorescent dans la macula des deux yeux
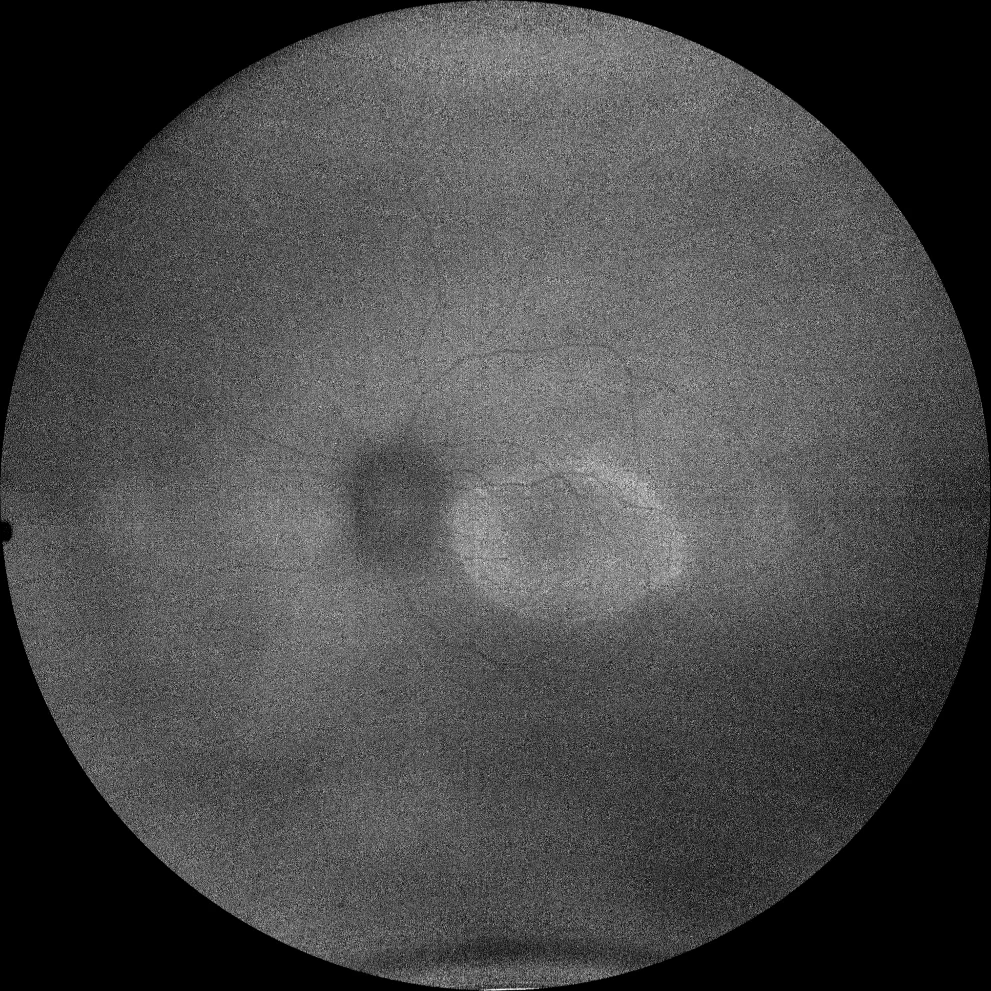
I et J. Autofluorescence (Clarus 500, Carl Zeiss Meditec ASG, Iéna, Allemagne) des yeux droit et gauche de la petite sœur, montrant l'anneau hyperautofluorescent dans la macula des deux yeux
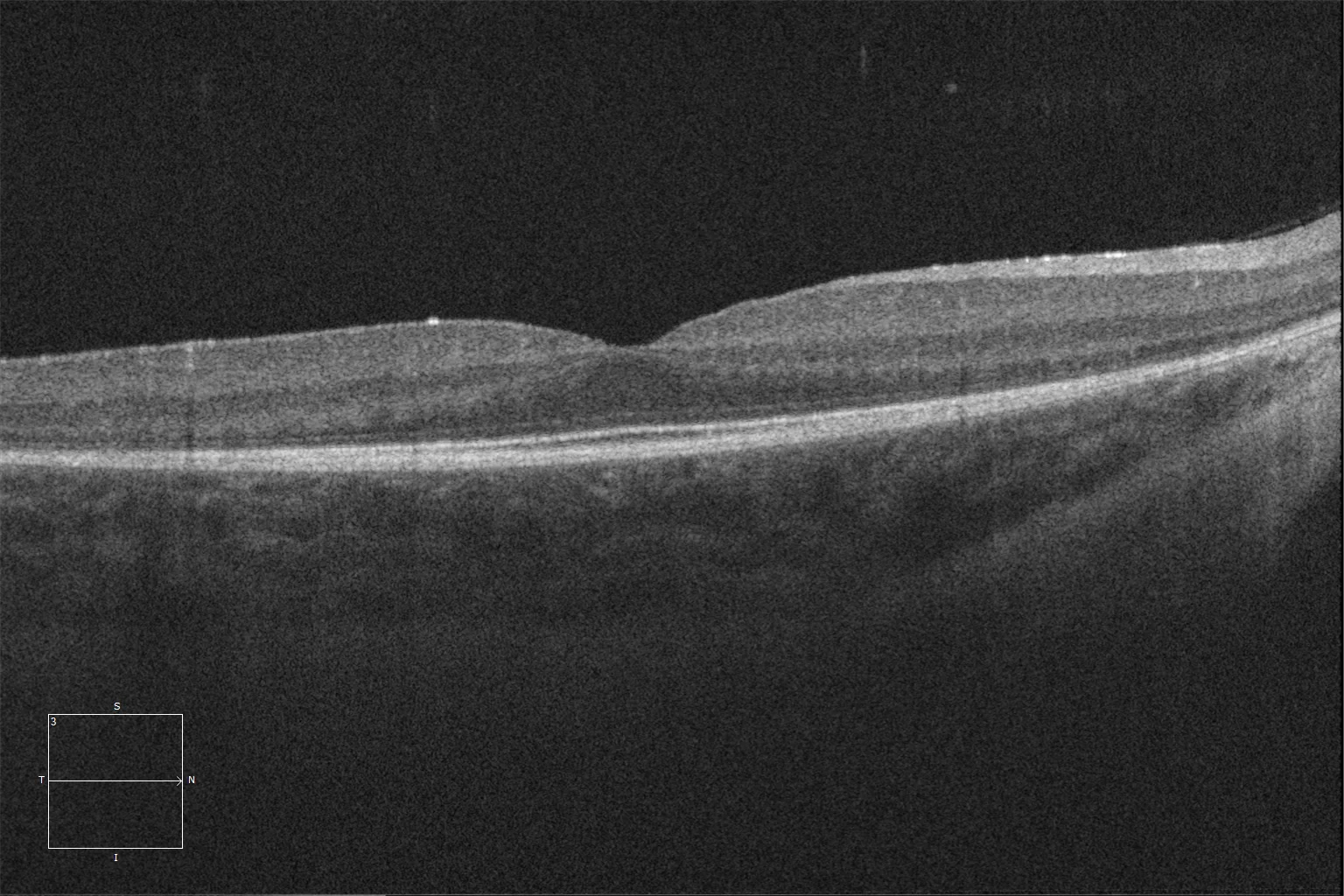
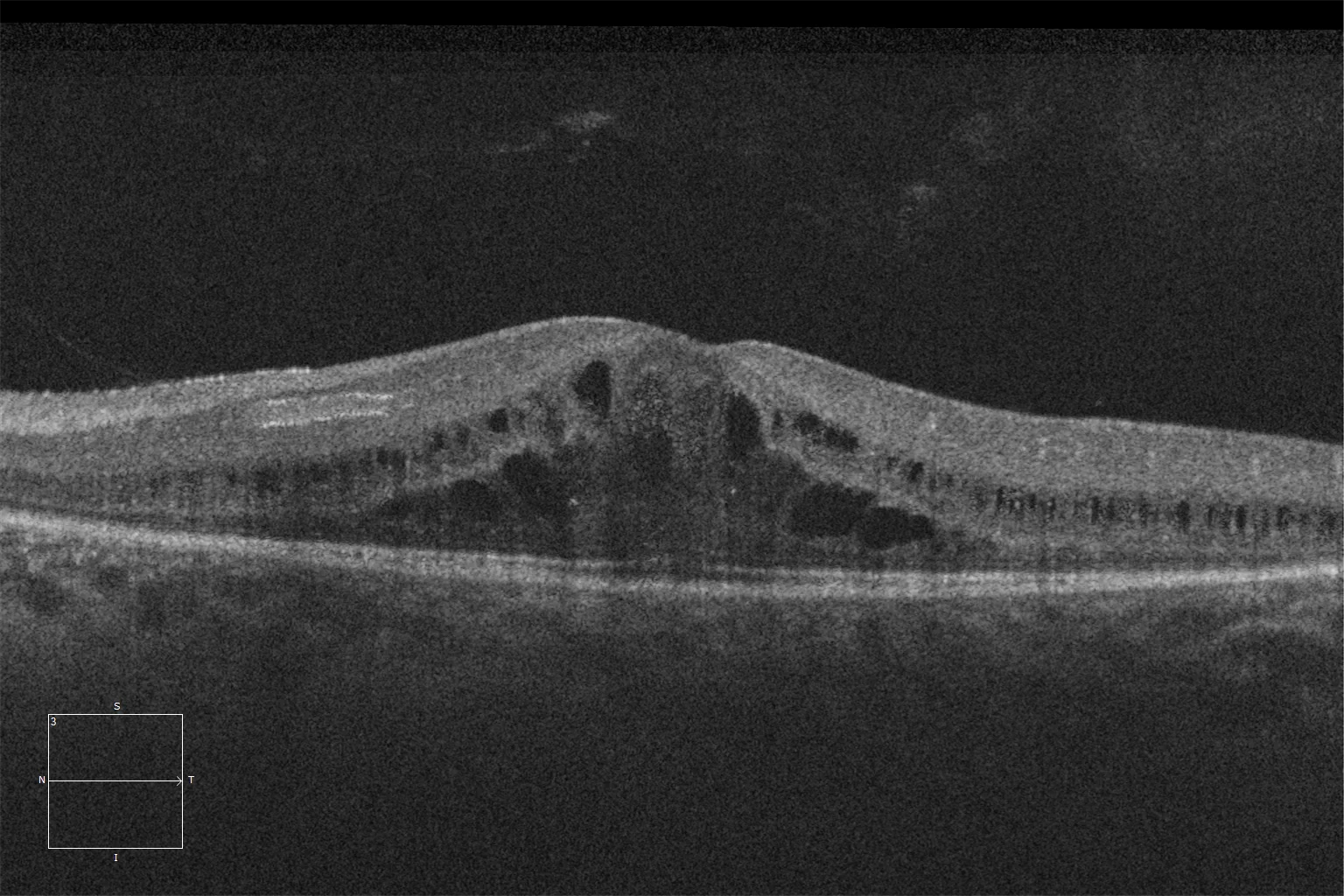
K et L. Tomographie par cohérence optique (Cirrus 5000, Carl Zeiss Meditec ASG, Jena, Allemagne) de la macula des yeux droit et gauche de la sœur cadette, montrant la préservation des couches ellipsoïde et ELM dans la rétine centrale et la perte de ces couches à mesure que l'on s'éloigne de la zone correspondant à l'anneau hyperautofluorescent.
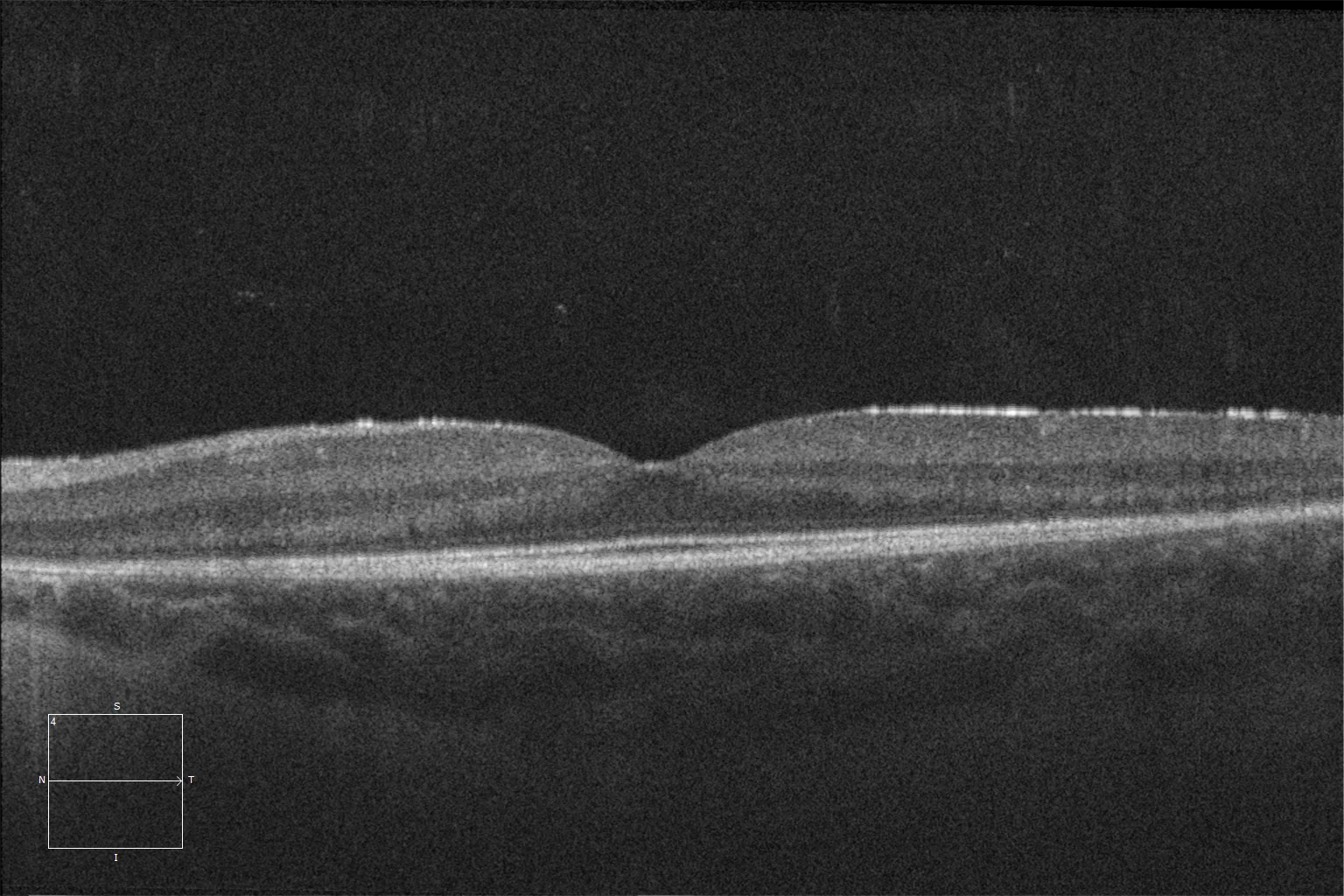
K et L. Tomographie par cohérence optique (Cirrus 5000, Carl Zeiss Meditec ASG, Jena, Allemagne) de la macula des yeux droit et gauche de la sœur cadette, montrant la préservation des couches ellipsoïde et ELM dans la rétine centrale et la perte de ces couches à mesure que l'on s'éloigne de la zone correspondant à l'anneau hyperautofluorescent.
Description
La rétinite pigmentaire (également appelée dystrophie des bâtonnets) regroupe un ensemble de maladies rétiniennes héréditaires, cliniquement et génétiquement hétérogènes. Elle se caractérise par un dysfonctionnement progressif de l’épithélium pigmentaire rétinien et des photorécepteurs, touchant initialement les bâtonnets puis les cônes. Elle se manifeste généralement par une baisse de la vision nocturne et une réduction du champ visuel périphérique, dès l’adolescence. La triade clinique classique comprend une atténuation antérieure, des anomalies pigmentaires rétiniennes en périphérie moyenne (généralement une hyperpigmentation sous forme de spicules osseux) et une pâleur cireuse de la papille rétinienne. Classiquement, l’électrorétinogramme (ERG) de champ complet révèle une diminution importante, voire une absence, des réponses scotopiques, ainsi qu’une diminution de la réponse photopique (qui peut être totalement absente aux stades avancés). Sa prévalence varie de 1 cas pour 3 000 à 1 cas pour 5 000. Elle peut survenir isolément – plus fréquemment – ou être associée à d’autres syndromes. Compte tenu de la grande variabilité génotypique, les modes de transmission autosomique dominant, autosomique récessif et récessif lié à l’X sont possibles. Même certaines maladies mitochondriales, comme le syndrome de Kearns-Sayre, peuvent se manifester par un phénotype de rétinite pigmentaire. Des cas isolés peuvent également survenir, parfois dus à des mutations de novo .